来看看能被我称为"神器"的,到底是什么?
APE:在R语言中分析系统发育和进化(在获得全基因组Alignment之后)
EmmanuelParadis,JulienClaudeKorbinianStrimmer
法国蒙彼利埃大学古生物生物学和系统发育实验室;慕尼黑大学统计学系

翻译APE发表的1篇原始文章,穿插一些代码、辅助信息,来了解APE的概况:

DOI:10.1093/bioinformatics/btg412
APE(AnalysisofPhylogeneticsandEvolution)是1个用R语言编写的用于分子进化和系统发育分析的免费软件包,提供了读写数据和操作系统发育树的实用函数,以及几种用于系统发育和进化分析的高级方法(比较和群体遗传方法)。
APE利用了许多用于统计和图形的R函数,并为开发、实现进一步的进化过程分析的统计方法,提供了灵活的框架。可从官方的R包存档(,现在可能为)获得。
从广义上说,系统发育分析涵盖了非常广泛的方法,从计算进化距离、重建基因树、估计分叉(Divergence)日期,到比较数据分析、进化速率估计和多样性(Diversification)分析。所有这些不同的任务都有一个特殊的共同点:严重依赖计算统计(Computationalstatistics)。R系统是一个免费的独立于平台的开源分析环境,最近(2004)已经形成统计计算和图形(Graphics)事实上的标准(IhakaGentleman,1996)。
R的1个优点是,通过编写专门的包,可以很容易地针对特定应用领域进行定制(Tailoredtoaparticularapplicationarea)。特别是,R在生物信息学中的有用性已经在基因表达数据(即转录组)的分析中得到了令人印象深刻的证明()。APE是首次在系统发育和进化数据的分析中发挥R的优势。APE专注于使用系统发育和系谱(Genealogical)树作为输入的统计分析。
在版中,APE提供了读取、写入、绘制、操作系统发育树,并在系统发育框架中分析比较数据(Comparativedata)、多样化分析、计算等位基因和核苷酸数据的距离、读取核苷酸序列和其它几个工具的功能,如Mantel测试、最小生成树的计算、群体遗传学参数的估计。
表1概述了APE中目前可用的函数。注意,有些方法(如比较法、天际线图/Skylineplot等)以前只能在专门的软件中使用。外部的树重建程序(如PHYLIP)可以通过标准的Shell命令从R调用。

表1.版中可用的特定函数
mst最小生成树-MinimumSpanningTree
phylo进化树在ape包中的数据对象的类(Class)名称-AsinglephylogenetictreemayhaveseveralrepresentationsintheNewickformatandinthe"phylo"classofobjectsusedin‘ape’.s溯祖.hivtree193个HIV-1序列的系统发育树数据.opsin视紫蛋白数据.
R语言的另1个优点是可以直接获得具有发表质量的图形输出,特别是使用PostScript设备时。例如,APE中的系统发育绘图功能处理颜色、线的粗细、字体、标签的间距,这些可为每个分枝(Branch)单独定义,因此可在单个系统发育图上表示3个不同的变量。
APE还产生复杂的群体遗传学图,如广义的天际线图(Generalizedskylineplot,Strimmer和Pybus,2001),只需1个命令。与任何R包一样,APE是命令行驱动的(重现、维护、共享非常方便)。函数由用户调用,可能带有参数和选项。在R中使用APE的任何会话(Session)都从此命令开始(当然,需要提前安装):
library(ape)
这使得APE的函数(执行特定的统计、绘图等功能)在R环境中可用。下面的命令可显示这些函数的列表(名称和简述):
library(help=ape)
APE中所有可用的R函数(表1)都以R超文本格式记录,有关它们使用的信息可以通过应用help命令检索,例如:
help()
以标准Newick插句格式保存在磁盘上的文本文件()中的进化树可被读取:
('')
这会将系统进化树存储在名为"tree1"的类(Class)'phylo'对象中。该对象中存储的信息(分枝长度)可通过输入`tree1`(命令)来检查,并且可通过执行以下(命令),来获得进化分枝图(Cladogram)形式的图形输出:
plot(tree1)
这实际上调用了APE的函数,绘制了系统发育树tree1。由于R的面向对象的性质,命令plot(x)(泛型函数?)可能会给出1个完全不同的结果,取决于对象所属的类(classofx)。默认情况下,树被绘制在图形窗口上(Graphicalwindow),但可以根据操作系统以各种文件格式导出。
除了这些简单的示例之外,在面向对象的结构中表示系统发育树可以直接操作进化分析中使用的各种计算(方法)的系统发育数据。目前在APE中实施的一些方法:系统发育独立对比(Felsenstein,1985;HarveyandPagel,1991),拟合生卒模型(Fittingbirth–,1994;PybusandHarvey,2000),群体遗传分析(Neeetal.,1995;StrimmerandPybus,2001),进化速率的非参数平滑(Sanderson,1997),利用Klastorin的方法在系统发育树中估计基因分组组(MisawaandTajima,2000)。
此外,在R函数hclust中实现的基于距离的聚类方法(Distancebasedclustering)可被APE使用,进而通过函数转换并生成类'phylo'类对象和'hclust'类对象。
APE中的类和方法(如'phylo')可以很容易地进一步扩展,来包括其它功能,例如:注释系统发育树。因此,APE不仅是一个数据分析包,也是一个开发和实现新方法的(开发)环境。此外,用C、C++或Fortran77编写的程序可从R中链接和调用,对于计算机密集型计算很有用。
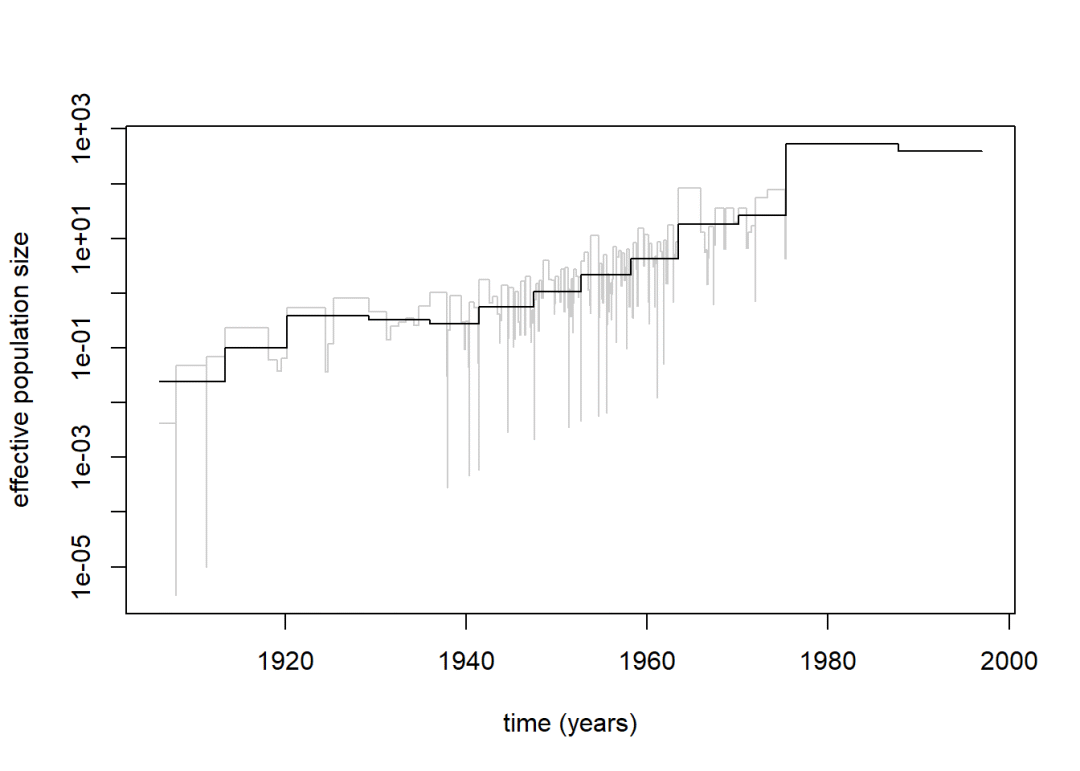
(附)APE绘制天际线图(示例,未美化)
gettreedata("")loadtreetoyrunremovecomments!!plotandcomparewithskylineplotsk-skyline()plot(sk,lwd=1,lty=3,=TRUE,=0.0023,=1997)lines(popsize,=TRUE,=0.0023,=1997)
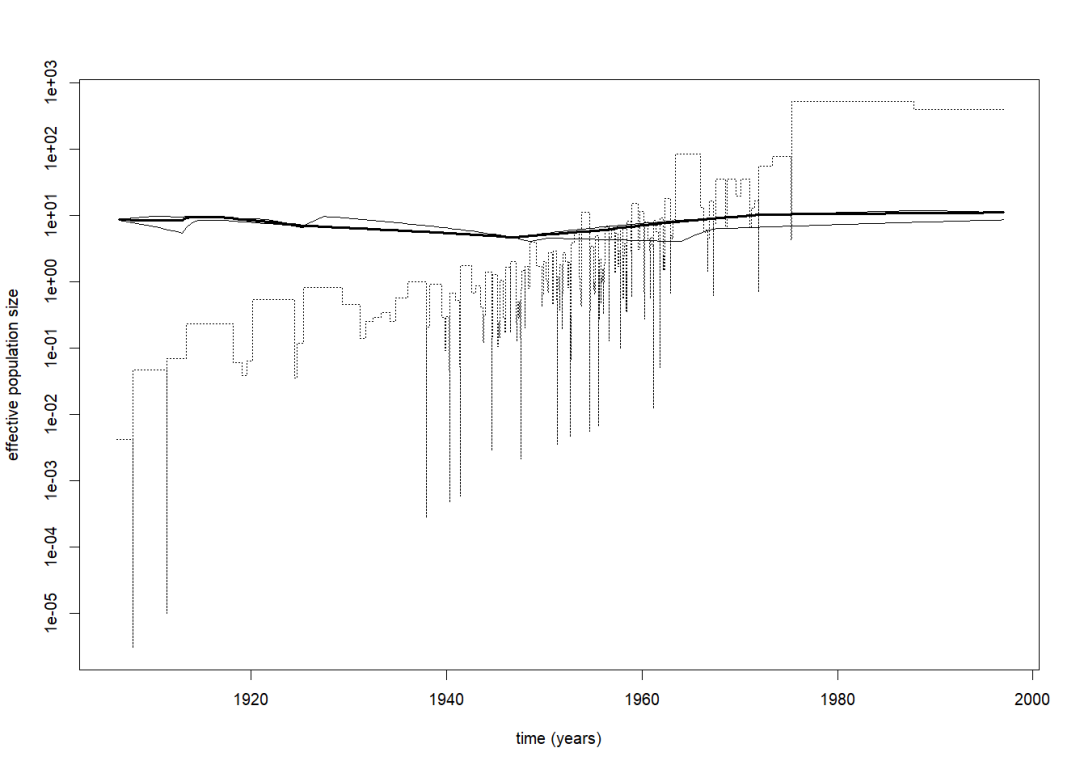
函数-可逆跳跃MCMC(马尔可夫链蒙特卡罗)推断群体数量史(DemographicHistory)
gettreedata("")loadtreefromtreefromcollapsedintervalssk1-skyline(ci)fromtreesk1plot(skyline())skylineplot()fromcollapsedintervalssk2-skyline(ci,0.0119)fromtreesk2plot(sk2)(ci)sk3-skyline(ci,-1)skylineplot函数-绘制天际线图(text=)sh()$epsilonclassicandgeneralizedskylineplotsk1-skyline()sk2-skyline(,0.0119)library(help=ape)
ape包的函数:
从文件中读取DNA序列,返回1个矩阵或DNA序列列表,分别将读取的分类群(Taxa)的名称作为rowname或names
序列以二进制格式返回(默认),或小写字母(选项=TRUE)
选项format:"fasta","sequential","clustal","interleaved"(序列名含空格时),或任何明确的缩写adegenet包的fasta2DNAbin函数:读取fasta格式的(序列)对齐,扩展名.此函数可提高内存效率,并且可以读取比Ape包的函数更大的数据集::fasta2DNAbin("./result/pairsnp/",quiet=FALSE,chunkSize=10,snpOnly=FALSE)
ConvertingFASTAalignmentintoaDNAbinobjectFindingthesizeofasinglegenomegenomesizeis:984nucleotides(2linespergenome)ImportingsequencesFormingfinalobjectdone.
str()其它函数:可下载、读取Fastq;;;;;clustal/muscle(多序列比对)dnds(,code=1,codonstart=1,quiet=FALSE,details=FALSE,=FALSE)data(woodmouse)res-dnds(woodmouse,quiet=TRUE)多序列比对图image(woodmouse)image(woodmouse,"n","blue")image(woodmouse,c("g","c"),"green")par(mfcol=c(2,2))barcodingstyle:image(woodmouse,x,"black",=0.5,=0.7)image(woodmouse[11:15,1:50],c("a","n"),c("blue","grey"))image(woodmouse,"g","yellow","black")phydataplot(x,phy,style="bars",offset=1,scaling=1,continuous=FALSE,width=NULL,leg="below",ring(x,phy,style="ring",offset=1,)(序列)标签管理:character、DNAbin、phylo、multiPhyloS3methodforclass'character'illegal="():;,[]",quote=FALSE,)makeLabel(x,tips=TRUE,nodes=TRUE,)makeLabel(x,tips=TRUE,nodes=TRUE,)makeLabel(x,)(‘’)((,model="raw"))plot(trw)
str(trw)
Listof4$edge:int[1:41,1:2]23252637372625272832$:num[1:41]6.78$:chr[1:22]"ERR2245277""ERR2245279""ERR2245288""ERR2245293"$Nnode:int20-attr(*,"class")=chr"phylo"-attr(*,"order")=chr"cladewise"
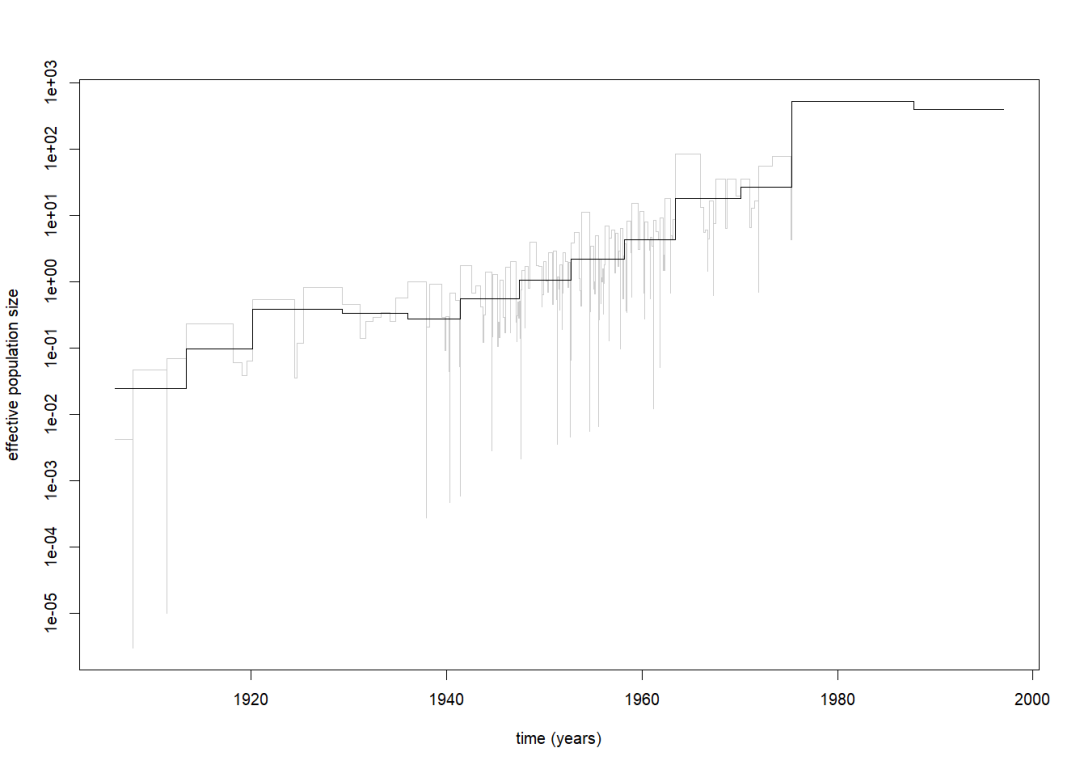
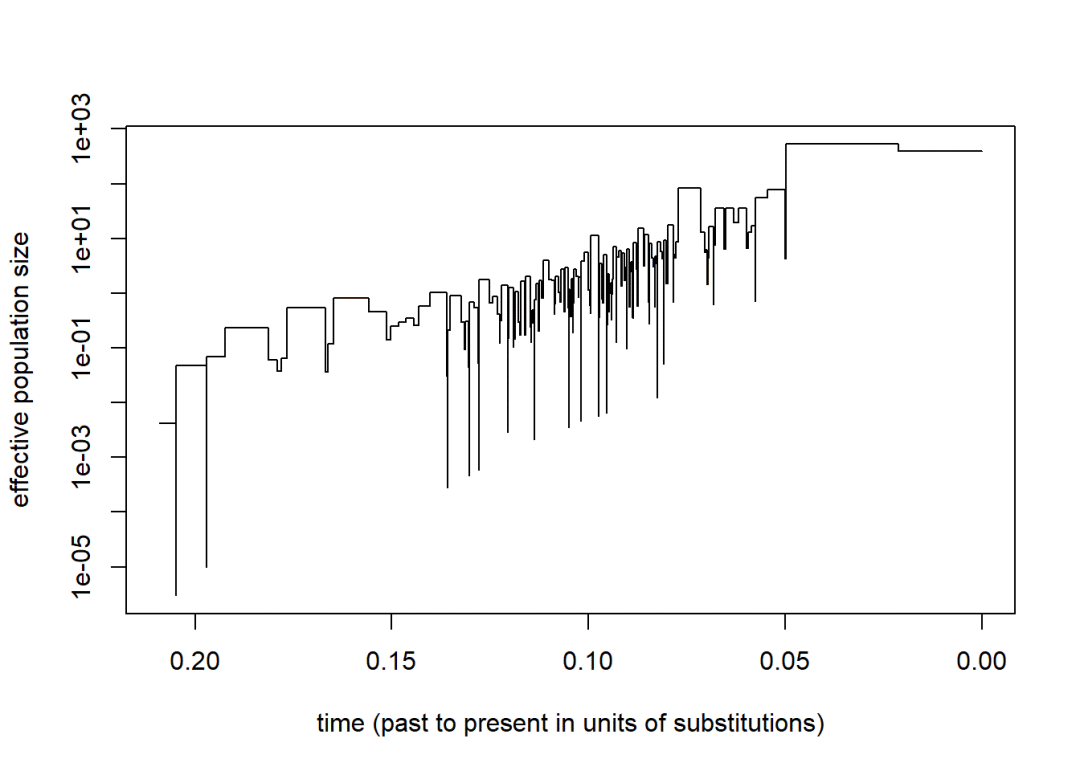
(trw,file="./",app=FALSE,digits=10,=FALSE)函数-可逆跳跃MCMC(马尔可夫链蒙特卡罗)推断群体数量史(DemographicHistory)(text=)(,nstep=100,thinning=1,=0,=FALSE)(,nstep=10000,thinning=5,=500)()skyline函数-使用天际线图估计有效种群大小(EffectivePopulationSize)(text=)()(ci,0)(ci,0.0119)classicskylineplotsk1-skyline(cl1)fromcoalescentintervalssk1-skyline()shortcut
plot(sk1,=TRUE,=0.0023,=1997)
generalizedskylineplotsk2-skyline(cl2)fromcoalescentintervalssk2-skyline(,0.0119)classicandgeneralizedskylineplottogetherinoneplotplot(sk1,=TRUE,=0.0023,=1997,col=c(grey(.8),1))lines(sk2,=TRUE,=0.0023,=1997)leg(.15,500,c("classic","generalized"),col=c(grey(.8),1),lty=1)
negativeepsilonalsotriggersestimationofepsilon
Searchingfortheoptimalepsilonepsilon=0.01191938
sk3$epsilon
[1]0.01191938
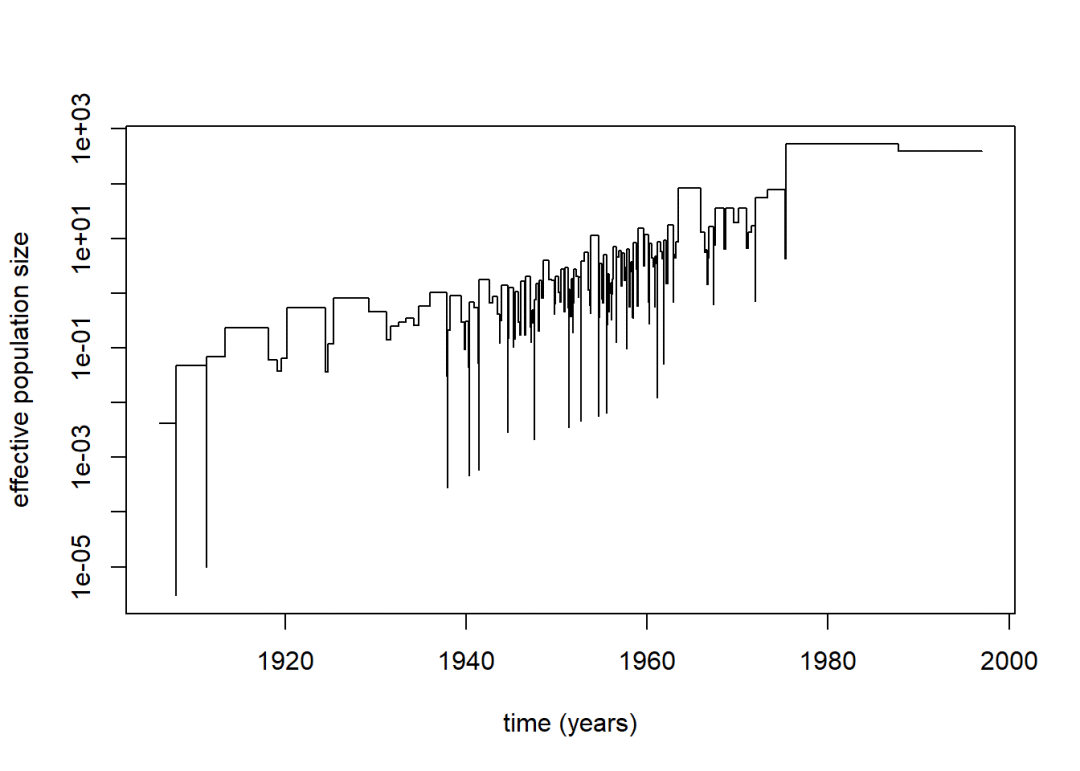
gettreedata("")loadtreeclassicskylineplotskylineplot()useyearsratherthansubstitutionsasunitforthetimeaxisplot(sk1,=TRUE,=0.0023,=1997,col=c(grey(.8),1))lines(sk2,=TRUE,=0.0023,=1997)leg(.15,500,c("classic","generalized"),col=c(grey(.8),1),lty=1)

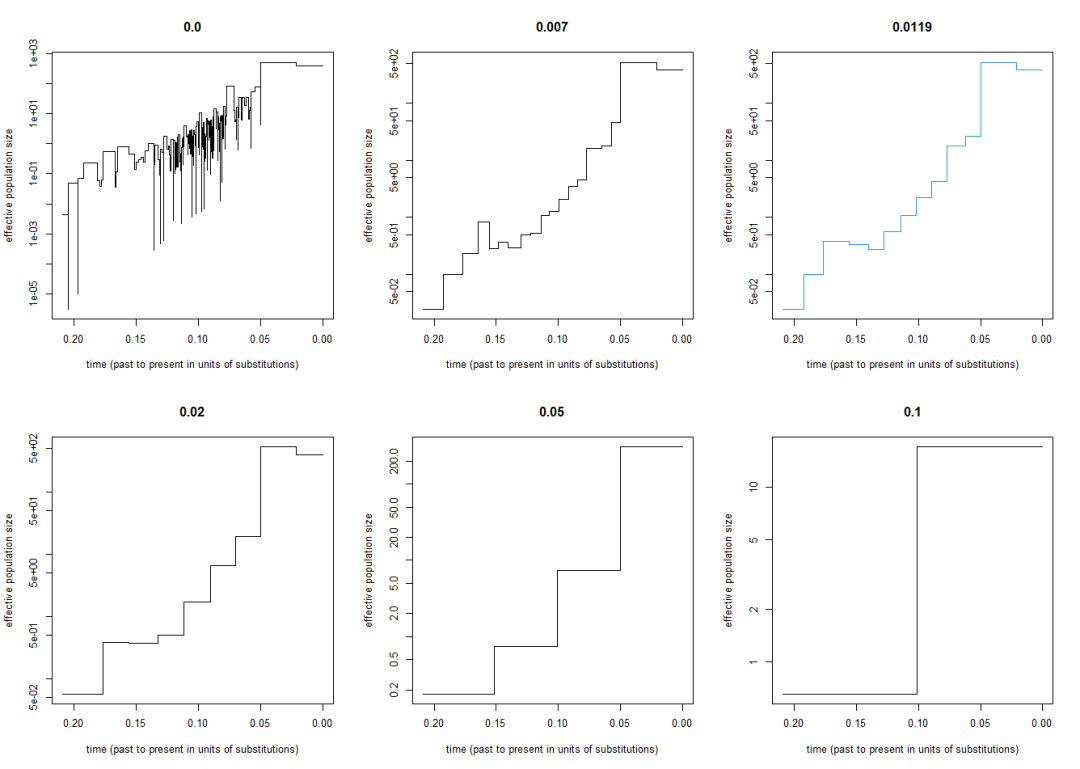
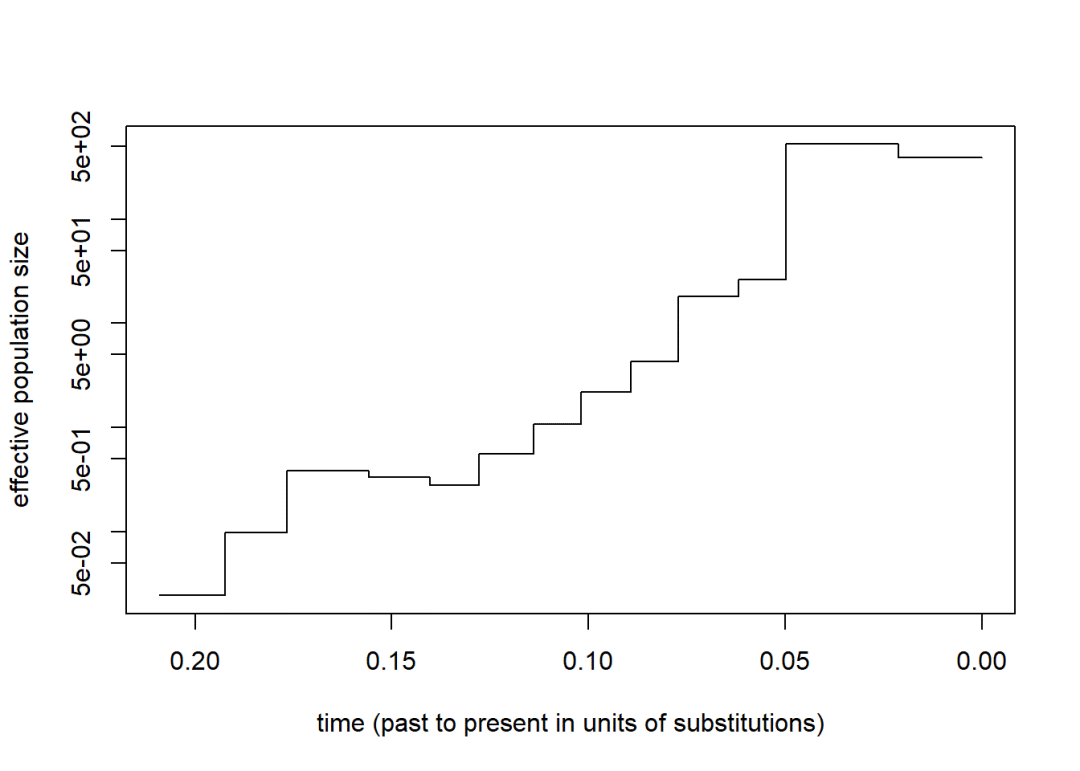
variousskylineplotsfordifferentepsilonslayout(mat=matrix(1:6,2,3,byrow=TRUE))()plot(skyline(ci,0.0));title(main="0.0")plot(skyline(ci,0.007));title(main="0.007")plot(skyline(ci,0.0119),col=4);title(main="0.0119")plot(skyline(ci,0.02));title(main="0.02")plot(skyline(ci,0.05));title(main="0.05")plot(skyline(ci,0.1));title(main="0.1")

layout(mat=matrix(1:1,1,1,byrow=TRUE)):对角线或方矩阵raw,N:每对序列之间差异位点的比例,或数量。有助于绘制“饱和图(Saturationplots)”。选项variance(方差)和gamma对其没有影响,但可以'dist'
'dist'num[1:231]727664115-attr(*,"Size")=int22-attr(*,"Labels")=chr[1:22]"ERR2245277""ERR2245279""ERR2245288""ERR2245293"-attr(*,"Upper")=logiFALSE-attr(*,"Diag")=logiFALSE-attr(*,"call")=languageape::(x=,model="N",variance=FALSE,gamma=FALSE,=FALSE,|__truncated__-attr(*,"method")=chr"N"
δ(,k=20,plot=TRUE,which=1:2)

TreeEstimationBasedonanImprovedVersionoftheNJAlgorithm,Gascuel(1997)trw-bionj(())trw-bionj((,model="N"))f-function(x)nj((x,model="raw"))for(iin1:5)TR-rmtree(100,10)length(pp10)andin100randomtreesof20labels?(TR)plot(pp10,pch="x",col=2)upgma()inphangorntree-fun()get100bootstraptrees:(tree,,fun,trees=TRUE)$trees
Runningbootstraps:100/100Calculatingbootstrapvaluesdone.
getproportionsofeachclade:(tree,bstrees,rooted=TRUE)getproportionsofeachbipartition:(tree,bstrees)layout(1)par(mar=rep(2,4))plot(tree,main="")drawSupportOnEdges(boot)nodelabels(clad)leg("bottomleft",leg=c("Bipartitions","Clades"),pch=22,=c("green","lightblue"),=2.5)

(,model="N")(d)
layout(1)(d,40,which=1)
(tr,"u")
(tr,"u"):39branchlength(s)NA(s):branchlengthsignoredintheplot
绘制带虚线、且右端对齐的树plot(trw,=0.1,=TRUE,font=1)(1)bootstrapwith100replications:((tw,,f,quiet=TRUE))
[1]NA234488901008923100100100100952610010029100100
thefirstvaluerelatestotherootnodeandisalways100itisignoredbelow:plot(tw,"u")tr-rtree(10)tr=tw绘制长枝打断的树plotBreakLongEdges(tr,2);axisPhylo()
plot(tr)
版权声明:本站所有作品(图文、音视频)均由用户自行上传分享,仅供网友学习交流,不声明或保证其内容的正确性,如发现本站有涉嫌抄袭侵权/违法违规的内容。请举报,一经查实,本站将立刻删除。